Stable Isotope Mixing Models in R with simmr
Andrew Parnell and Richard Inger
2023-10-27
Source:vignettes/simmr.Rmd
simmr.RmdIntroduction
tl;dr see the Quick Start vignette
simmr is a package designed to solve mixing equations
for stable isotopic data within a Bayesian framework. This guide is
designed to get researchers up and running with the package and giving
them a full list of all the available features. No expertise is required
in the use of R.
simmr is designed as an upgrade to the SIAR package and
contains many of the same features. This new version contains a slightly
more sophisticated mixing model, a simpler user interface, and more
advanced plotting features. The key differences between SIAR and
simmr are:
-
simmrhas a slightly richer mixing model based on code from the Parnell et al 2013 Environmetrics paper -
simmrdoes not have a menu system; all commands must be run through the console or script windows -
simmruses ggplot2 to create graphs and JAGS to run the mixing model
We assume that you have a sound working knowledge of stable isotopic mixing models, and the assumptions and potential pitfalls associated with these models. A list of required reading is presented in Appendix A of this guide. We strongly recommend reading Philips et al 2015, Canadian Journal of Zoology for the basic assumptions and dos and don’ts of running mixing models.
We assume that if you have got this far you have installed R. We also recommend
installing Rstudio as this provides a
very neat interface to use R and simmr. The instructions
below all assume you are using Rstudio.
If you find bugs in the software, or wish to suggest new features,
please add your input to the simmr GitHub issues
page.
Installation of the simmr package
The simmr package uses the JAGS (Just Another Gibbs
Sampler) programmer to run the stable isotope mixing model. Before you
install simmr, visit the JAGS website and
download and install JAGS for your operating system.
Next, start Rstudio and find the window with the command prompt (the
symbol >). Type
install.packages("simmr")It may ask you to pick your nearest CRAN mirror (the nearest site
which hosts R packages). You will then see some activity on the screen
as the simmr package and the other packages it uses are
downloaded. The final line should then read:
package 'simmr' successfully unpacked and MD5 sums checked
You then need to load the package. Type
This will load the simmr package and all the associated
packages. You’ll need to type the library(simmr) command
every time you start R. If you haven’t installed JAGS properly you will
be informed at this point.
Considerations before running simmr
Before getting started there are a couple of points to consider.
Working with scripts
The best way to use the simmr package is by creating
scripts. A script can be created in Rstudio by clicking
File > New File > Rscript. This opens a text window
which allows commands to be typed in order and saved. The command can be
sent to the command prompt (which Rstudio calls the Console) by
highlighting the command and clicking Run (or going to Code > Run
Lines). There are also keyboard shortcuts to speed up the process. We
strongly recommend you learn to run R via scripts.
Data Structure
simmr can handle three different types of data
structure:
- A single consumer. This may occur when you have only one data point on a single individual
- Multiple consumers. This may occur if you have multiple individuals in a single sampling period
- Multiple groups of consumers. This may occur if you have multiple consumers which are observed over different sampling periods/locations, different demographic groups, etc.
Unless you specify a grouping variable simmr assumes
that all the observations are from the same group. If you have extra
variables (e.g. explanatory variables) that you think may influence the
dietary proportions, you should consider using MixSIAR instead.
How to run simmr
The general structure for running simmr is as
follows:
- Call
simmr_loadon the data to get it into the right format - Plot the data in isotope space (‘iso-space’) using
plot - Run the mixing model with
simmr_mcmcorsimmr_ffvb - Check the model converged with
summary - Check the model fit is calibrated with
posterior_predictive - Explore the results with
plotandsummary, andprior_viz. If you have multiple groups and want to compare output between them, use the `compare_groups’ function
For the next part of this document, we concentrate on simple examples without grouping structure.
Step 1: Getting the data into simmr
simmr requires at minimum 3 input objects; the consumers
or mixtures, the source means, and the source
standard deviations. Optionally, you can also add correction data
(also called trophic enrichment factors, TEFs) represented again as
means and standard deviations, and concentration dependence values. The
easiest way to get data into simmr is to create an Excel file, as shown
in the included vignette('quick_start) guide. Alternatively
you can copy and past your data, comma separated, as below:
mix <- matrix(c(
-10.13, -10.72, -11.39, -11.18, -10.81, -10.7, -10.54,
-10.48, -9.93, -9.37, 11.59, 11.01, 10.59, 10.97, 11.52, 11.89,
11.73, 10.89, 11.05, 12.3
), ncol = 2, nrow = 10)
colnames(mix) <- c("d13C", "d15N")
s_names <- c("Zostera", "Grass", "U.lactuca", "Enteromorpha")
s_means <- matrix(c(-14, -15.1, -11.03, -14.44, 3.06, 7.05, 13.72, 5.96), ncol = 2, nrow = 4)
s_sds <- matrix(c(0.48, 0.38, 0.48, 0.43, 0.46, 0.39, 0.42, 0.48), ncol = 2, nrow = 4)
c_means <- matrix(c(2.63, 1.59, 3.41, 3.04, 3.28, 2.34, 2.14, 2.36), ncol = 2, nrow = 4)
c_sds <- matrix(c(0.41, 0.44, 0.34, 0.46, 0.46, 0.48, 0.46, 0.66), ncol = 2, nrow = 4)
conc <- matrix(c(0.02, 0.1, 0.12, 0.04, 0.02, 0.1, 0.09, 0.05), ncol = 2, nrow = 4)The mix object above contains the stable isotopic data
for the consumers. The data should be listed as the consumer values for
the first isotope, followed by the consumer values for the second
isotope and so on. The matrix function turns this into a
matrix (a rectangle of numbers) with 2 columns. The first column
contains the data for isotope 1, and the second the data for isotope 2.
Any number of isotopes and observations can be used. It is recommended
but not necessary to give the mixtures column names representing the
isotopes to which each column corresponds.
The source names are provided in the s_names object, and
the source means and standard deviations in s_means and
s_sds. These latter objects must also be matrices, where
the number of rows is the number of sources, and the number of columns
the number of isotopes. In each case, the data are included by listing
the values for the first isotope, then the second isotope, and so
on.
The correction data is stored in c_means and
c_sds. Again this should be a matrix of the same dimension
as s_means and s_sds. Finally the
concentration dependencies (i.e. the elemental concentration values) are
included as conc.
Some data sets are also included in simmr for quick
access to examples. See data(package = "simmr") for the
list. They can all be accessed via,
e.g. data("geese_data").
To load the data into simmr, use:
simmr_in <- simmr_load(
mixtures = mix,
source_names = s_names,
source_means = s_means,
source_sds = s_sds,
correction_means = c_means,
correction_sds = c_sds,
concentration_means = conc
)Remember that the correction_means,
correction_sds, and concentration_means are
optional.
Step 2: Plotting the data in iso-space
We can now plot the raw isotopic data with:
plot(simmr_in)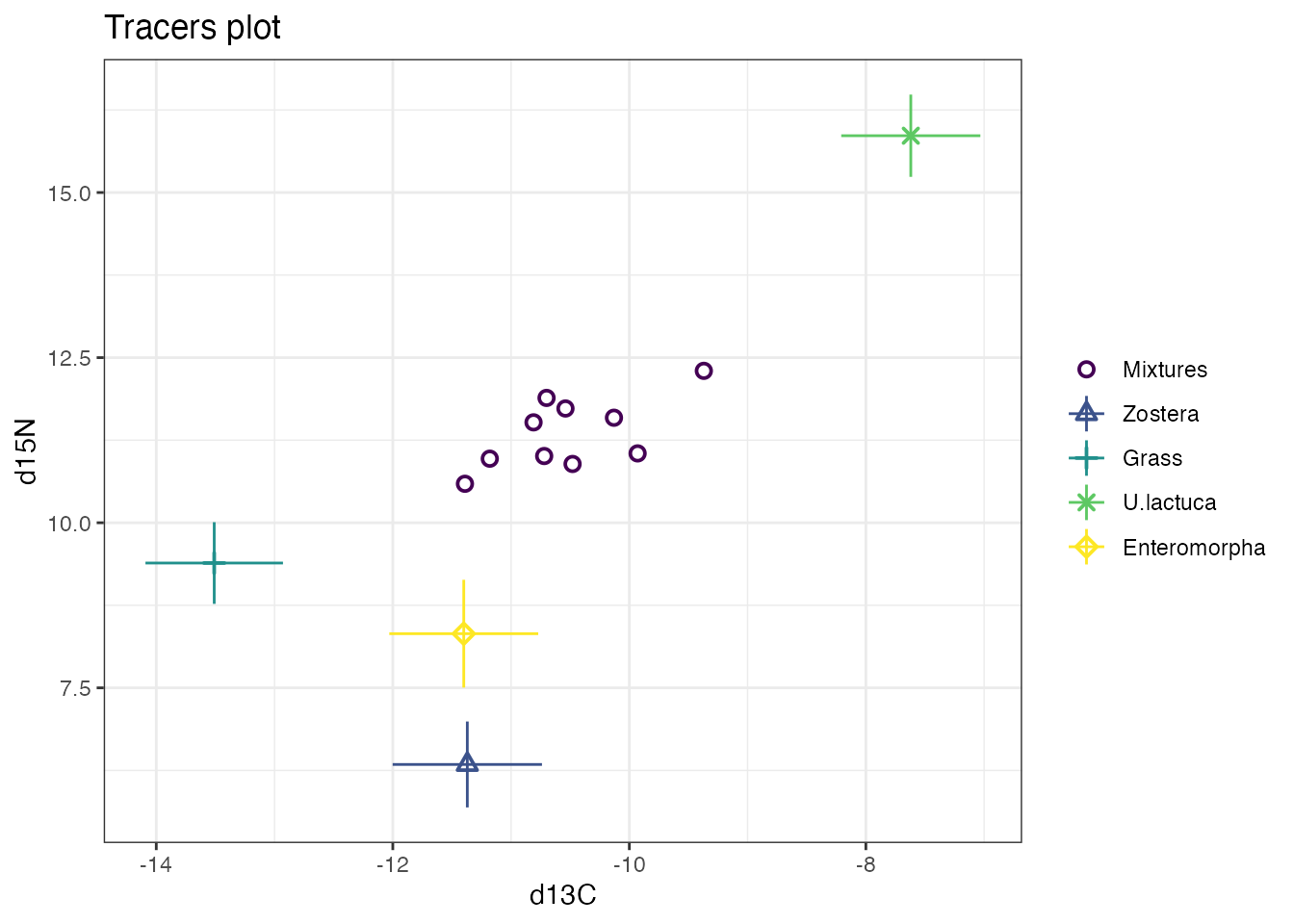
This will produce a biplot with the isotope that is in the first column on the x-axis, and the isotope in the second column on the y-axis. You can make the plot slightly nicer with some extra arguments:
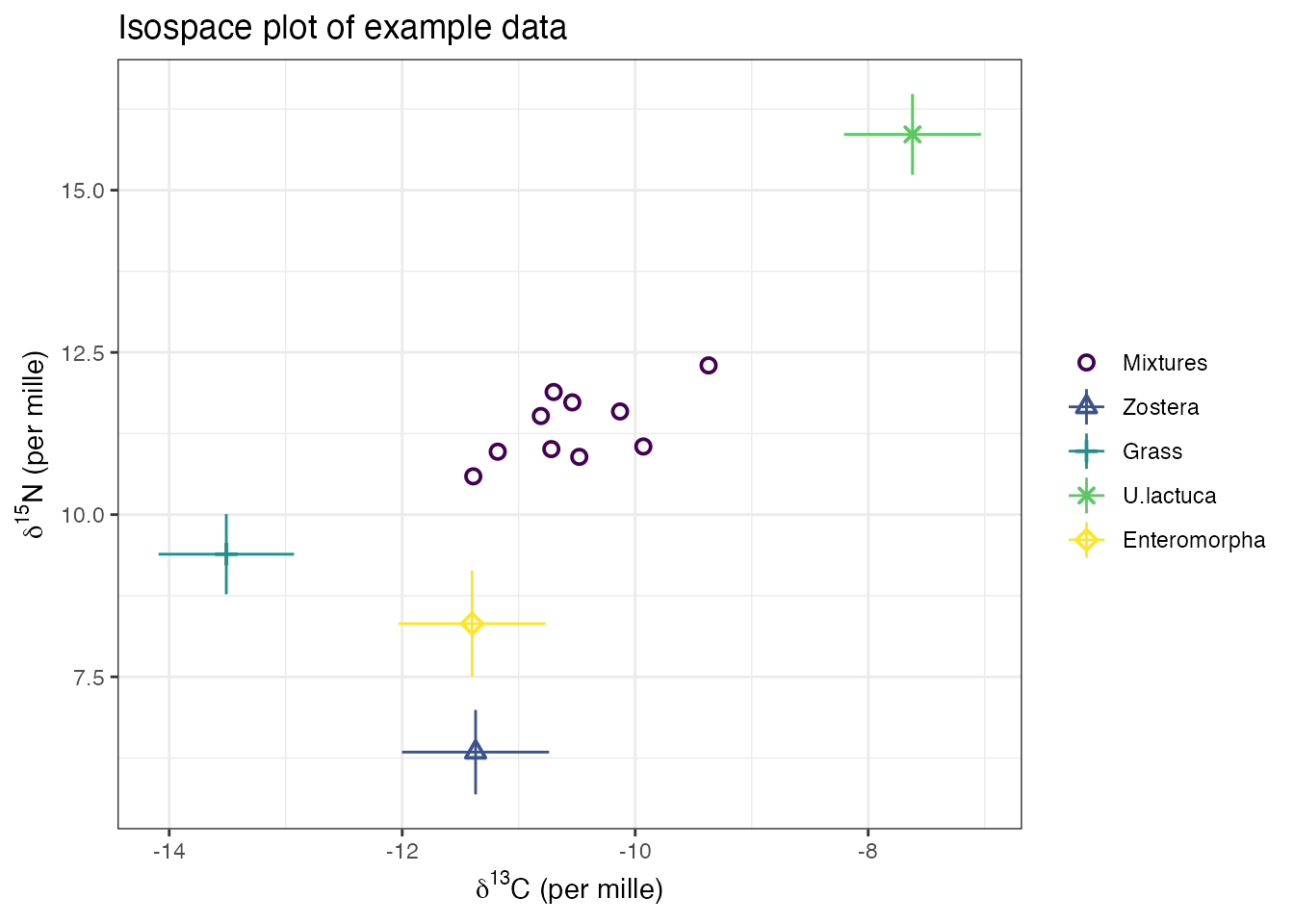
plot(simmr_in,
xlab = expression(paste(delta^13, "C (per mille)",
sep = ""
)),
ylab = expression(paste(delta^15, "N (per mille)",
sep = ""
)),
title = "Isospace plot of example data"
)
See the help file help(plot.simmr_input) for more
options on the plotting commands, including the ability to plot
different tracers/isotopes when there are more than 2 isotopes.
If all the mixtures lie inside the mixing polygon defined by the
sources, then the data are acceptable for running simmr.
See Philips et al 2015, Canadian Journal of Zoology for more details on
when data are suitable for running through a mixing model.
Step 3: Running simmr
The next step is to actually run the model. There are two options to choose from here. The code to run the model using a Markov chain Monte Carlo (MCMC) algorithm is as follows:
simmr_out <- simmr_mcmc(simmr_in)## module glm loadedThis command takes the object simmr_in we created
earlier and uses it as input for the model. It tells simmr
to store the output from the model run in an object called
simmr_out.
Alternatively, it can be run using a Fixed Form Variational Bayes (FFVB) algorithm, as follows:
simmr_out_ffvb <- simmr_ffvb(simmr_in)The model should take less than a minute to run, though this will depend on the speed of the computer you are using. Other data sets might take slightly longer or shorter depending on the number of sources, isotopes, and observations. The progress of the model is displayed on the command line window, which shows the percentage complete.
Step 4: Checking the algorithm converged
Markov chain Monte Carlo (MCMC) works by repeatedly guessing the
values of the dietary proportions and find those values which fit the
data best. The initial guesses are usually poor and are discarded as
part of an initial phase known as the burn-in. Subsequent iterations are
then stored and used for the posterior distribution; the best
estimates of the dietary proportions given the data and the model.
Because it can take many thousands of iterations to move away from the
initial guesses, convergence diagnostics can be created to
check the model has run properly. In simmr this is done
with:
summary(simmr_out, type = "diagnostics")##
## Summary for 1## R-hat values - these values should all be close to 1.## If not, try a longer run of simmr_mcmc.## deviance Zostera Grass U.lactuca Enteromorpha sd[d13C]
## 1 1 1 1 1 1
## sd[d15N]
## 1If the model run has converged properly the values should be close to
1. If they are above 1.1, we recommend a longer run. See
help(simmr_mcmc) for how to do this.
Fixed Form Variational Bayes (FFVB) is an optimisation based technique. It doesn’t require this diagnostic function.
Step 5: Checking the model fit
You can check the fit of the model with a posterior predictive check. This is similar to a fitted values plot in a linear regression. If the data points (denoted by the plot as \(y\)) broadly lie in the fitted value intervals (denoted \(y_rep\); the default is a 50% interval) then the model is fitting well:
post_pred <- posterior_predictive(simmr_out)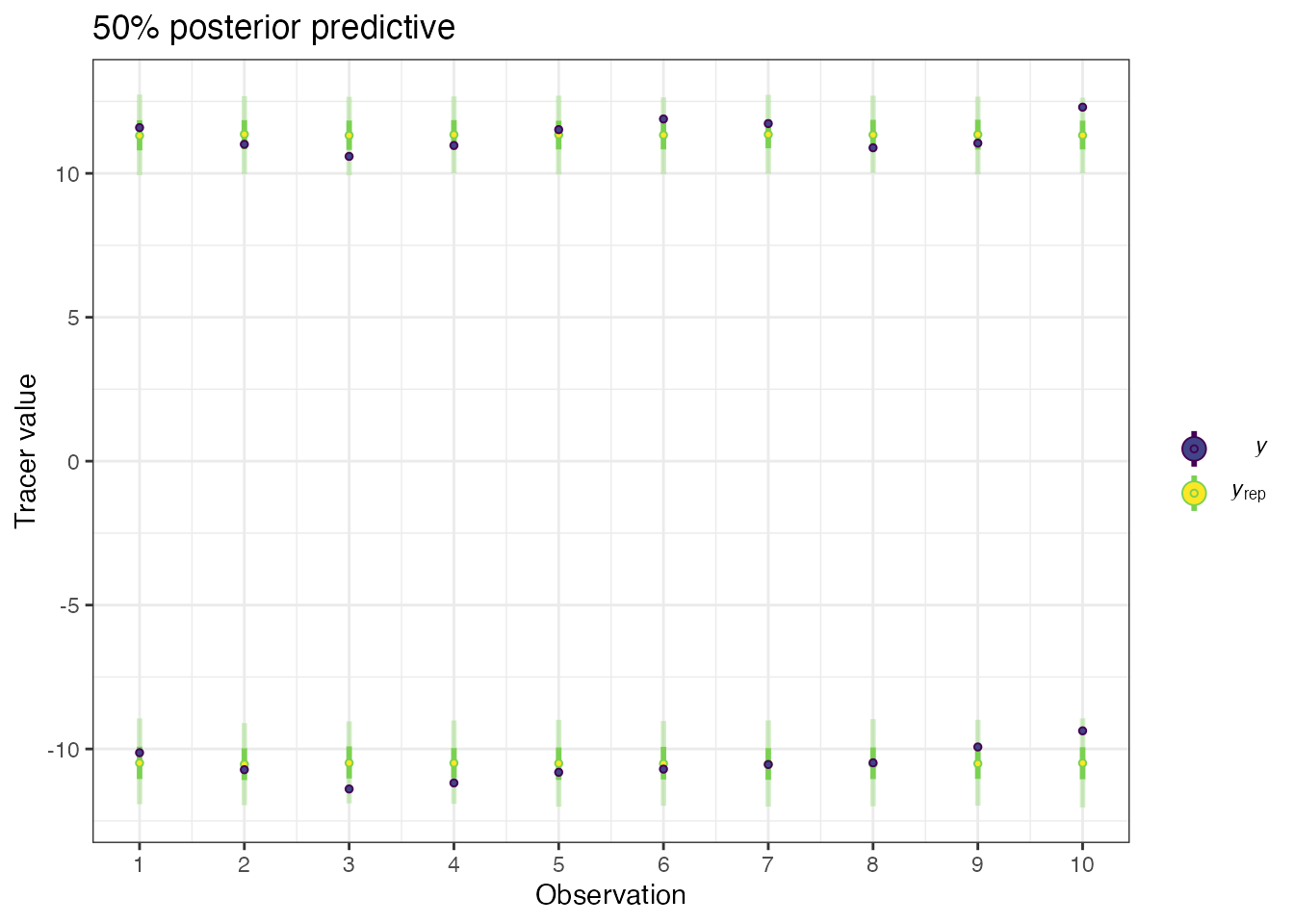
print(post_pred)The output includes a table showing which observations lie outside
the posterior predictive and the proportion doing so, which should
approximately match the proportion specified in the
posterior_predictive function (default 50%).
Step 6: Exploring the results
All SIMMs use informative (usually generalist) prior distributions as a default. These functions work whether the model has been run through MCMC or FFVB. You can plot the priors and the posteriors with:
prior_viz(simmr_out)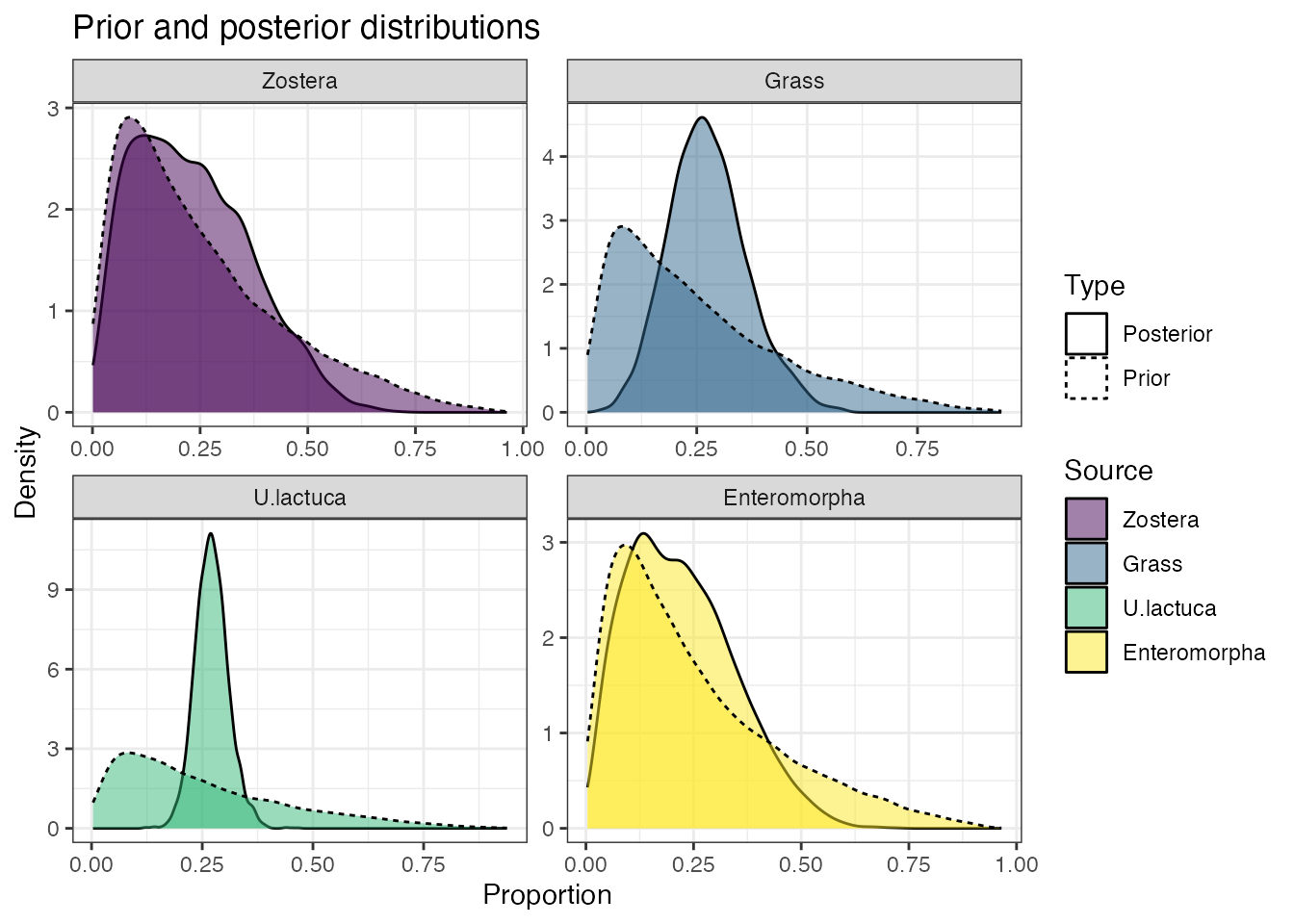
simmr produces both textual and graphical summaries of
the model run. Starting with the textual summaries, we can get tables of
the means, standard deviations and credible intervals (the Bayesian
equivalent of a confidence interval) with:
summary(simmr_out, type = "statistics")##
## Summary for 1## mean sd
## deviance 39.278 4.950
## Zostera 0.232 0.134
## Grass 0.275 0.088
## U.lactuca 0.272 0.037
## Enteromorpha 0.222 0.123
## sd[d13C] 0.743 0.301
## sd[d15N] 0.664 0.299
summary(simmr_out, type = "quantiles")##
## Summary for 1## 2.5% 25% 50% 75% 97.5%
## deviance 32.695 35.599 38.327 41.908 51.477
## Zostera 0.033 0.122 0.216 0.326 0.516
## Grass 0.118 0.214 0.270 0.330 0.467
## U.lactuca 0.202 0.246 0.270 0.295 0.351
## Enteromorpha 0.036 0.124 0.209 0.305 0.492
## sd[d13C] 0.328 0.541 0.689 0.885 1.446
## sd[d15N] 0.244 0.463 0.612 0.797 1.406These suggest that the dietary proportions for this model are quite uncertain. However we can see that the credible interval for U.lactuca is the narrowest, running from approximately 20% to 35% of the diet. The reason this one is the narrowest can be seen from the isospace plot - this source is the most clearly separated from the others.
simmr can also produce histograms, boxplots, density
plots, and matrix plots of the output. Starting with the density
plot:
plot(simmr_out, type = "density")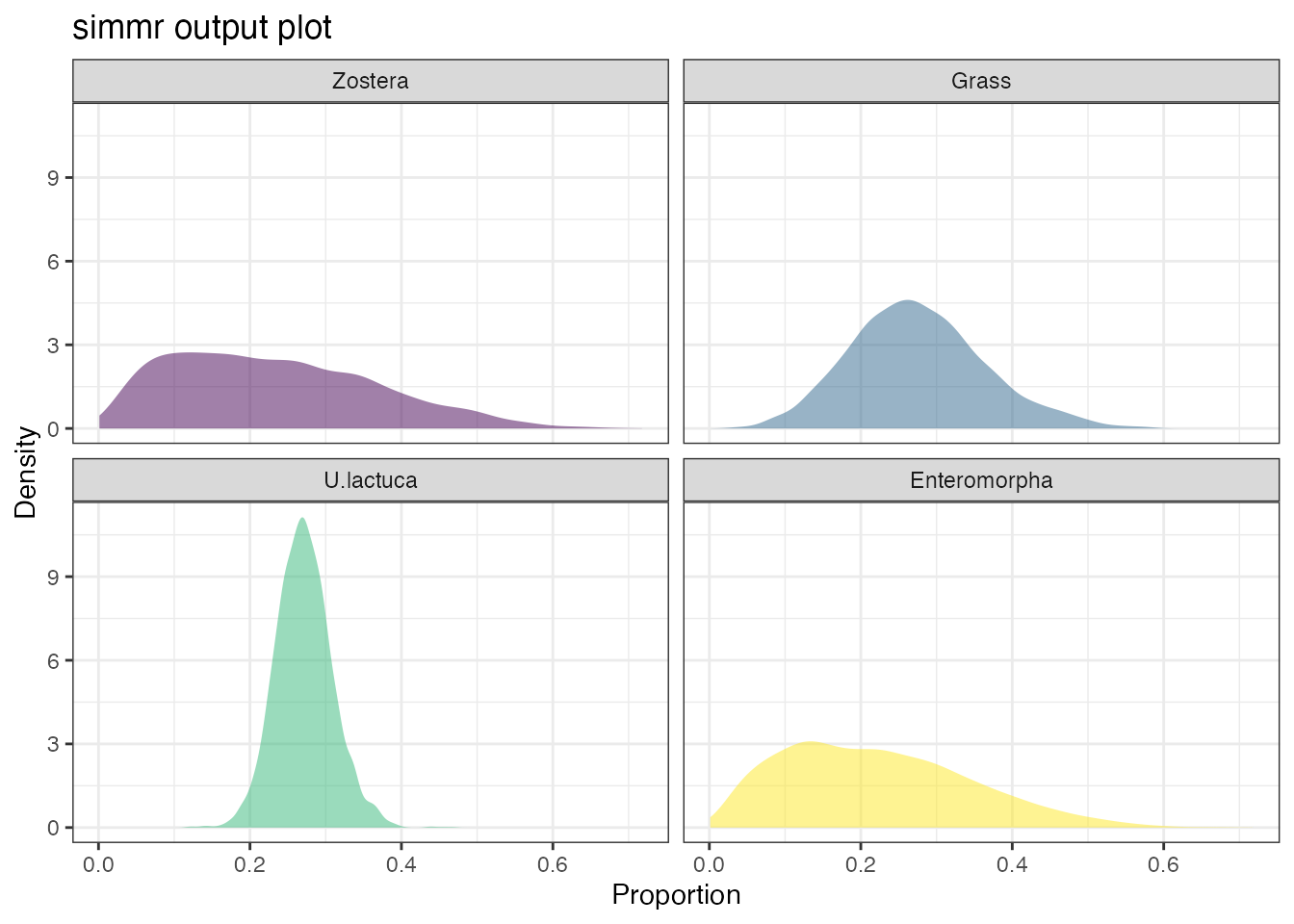
We can see that Zostera and Enteromorpha are poorly constrained in comparison to Grass and especially U.lactuca. Again this is unsurprising since the isospace plot indicated that these were the two most clearly separated sources.
The most useful output plot is the matrix plot:
plot(simmr_out, type = "matrix")## Registered S3 method overwritten by 'GGally':
## method from
## +.gg ggplot2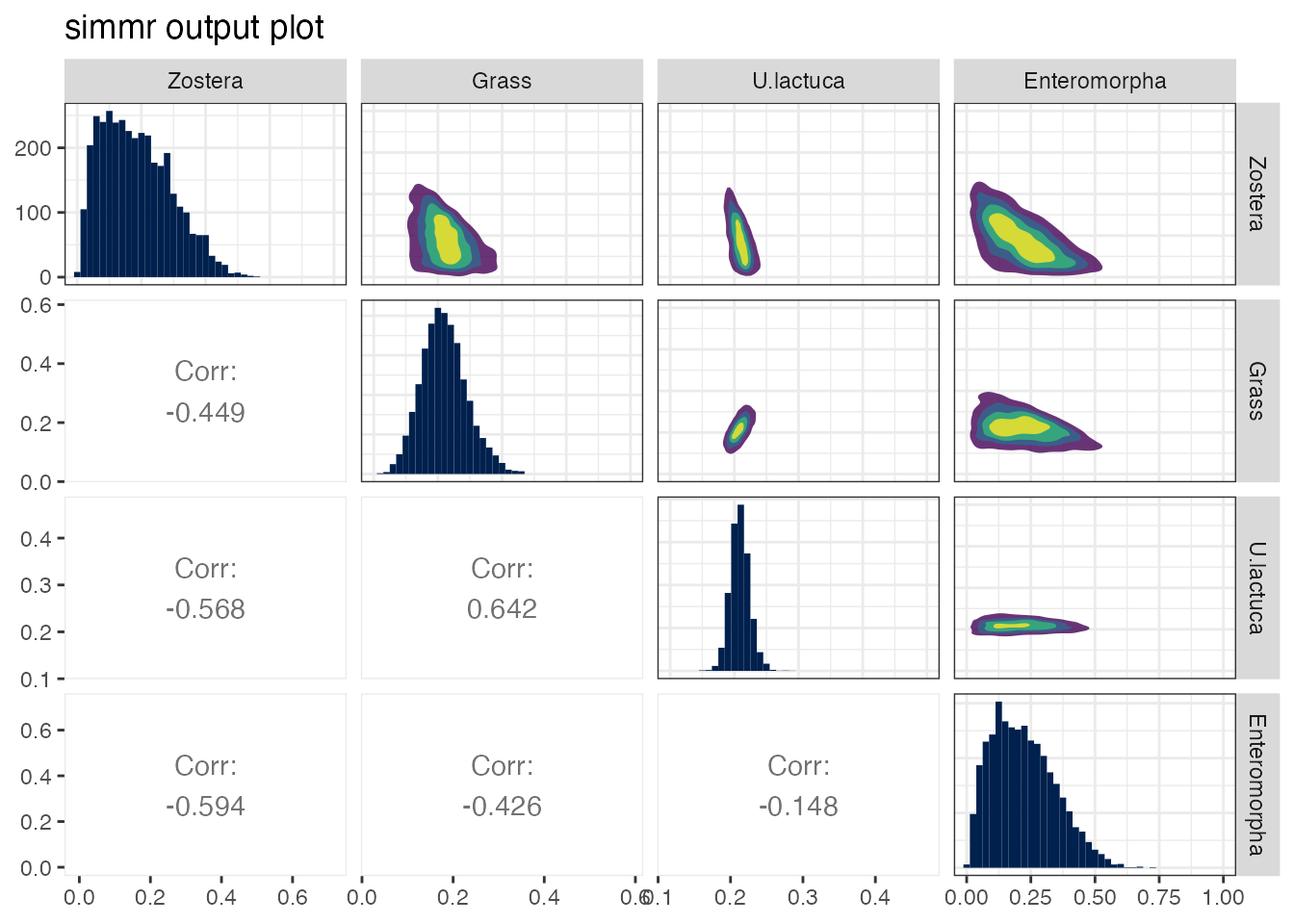
This shows the source histograms on the diagonal, contour plots of the relationship between the sources on the upper diagonal, and the correlation between the sources on the lower diagonal. Large negative correlations indicate that the model cannot discern between the two sources; they may lie close together in iso-space. Large positive correlations are also possible when mixture data lie in a polygon consisting of multiple competing sources. Here the largest negative correlation is between Zostera and Enteromorpha. This is because they lie closest together in isospace. In general, high correlations (negative or positive) are indicative of the model being unable to determine which food sources are being consumed, and are an unavoidable part of stable isotope mixing models.
If you want to compare the dietary proportions between two different
sources, you can use the compare_sources function. This
takes two or more sources and compares the dietary proportions with an
optional plot. For example:
compare_sources(simmr_out,
source_names = c("Zostera", "U.lactuca")
)## Prob (proportion of Zostera > proportion of U.lactuca) = 0.376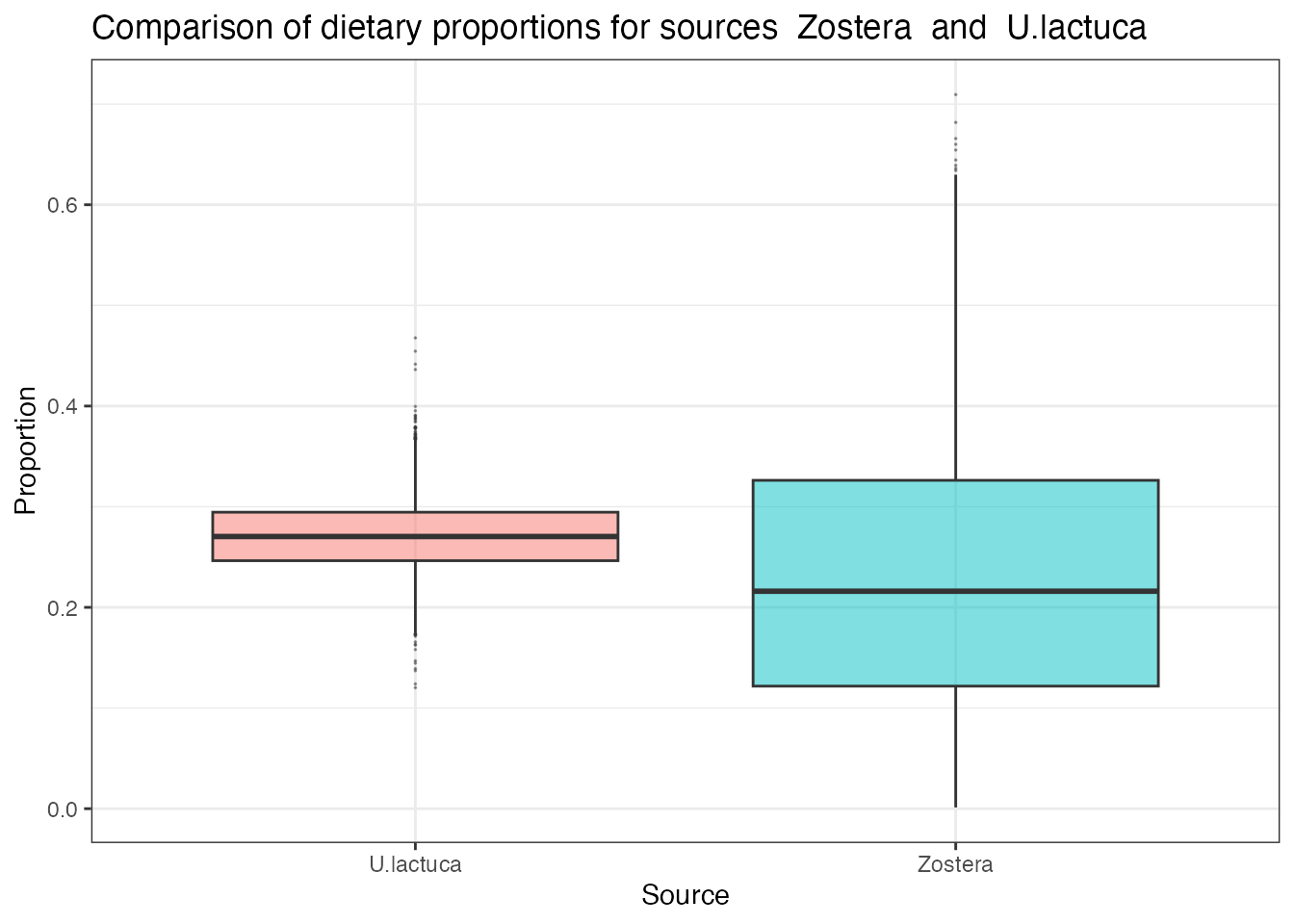
This produces a direct probability that the dietary proportion for the first source is bigger than that of the second. If you want to compare more than two sources, specify them with:
compare_sources(simmr_out,
source_names = c(
"Zostera",
"U.lactuca",
"Enteromorpha"
)
)## Most popular orderings are as follows:## Probability
## Zostera > U.lactuca > Enteromorpha 0.3031
## Enteromorpha > U.lactuca > Zostera 0.2594
## U.lactuca > Enteromorpha > Zostera 0.1958
## U.lactuca > Zostera > Enteromorpha 0.1689
## Zostera > Enteromorpha > U.lactuca 0.0386
## Enteromorpha > Zostera > U.lactuca 0.0342
For further information and options on comparing sources, see
help(compare_sources).
How to run simmr on multiple groups
In many cases we have data from different sampling locations, or
different types of individuals (e.g. male/female) and we to compare
between these groups. simmr can handle these data sets
provided they all share the same sources, corrections and concentration
dependence values.
A useful data set is given by Inger et al 2006 and provided as part of the original SIAR package. These data concern Brent Geese observed on 8 separate sampling periods.
These data are included in the package, and we can load these data into R with:
data(geese_data)…and into simmr with:
simmr_groups <- with(
geese_data,
simmr_load(
mixtures = mixtures,
source_names = source_names,
source_means = source_means,
source_sds = source_sds,
correction_means = correction_means,
correction_sds = correction_sds,
concentration_means = concentration_means,
group = groups
)
)Note that the group object above is specified to be a
factor but can also be an integer, the levels of which will appear in
plots. However, when specifying groups in later commands you should use
the integer values to reference which groups to plot
Next it is a matter of following the simmr commands as
before to load in, with an extra argument specifying the groups:
When we create the isospace plot we can specify which groups we wish to plot:
plot(simmr_groups,
group = 1:8,
xlab = expression(paste(delta^13, "C (per mille)",
sep = ""
)),
ylab = expression(paste(delta^15, "N (per mille)",
sep = ""
)),
title = "Isospace plot of Inger et al Geese data",
mix_name = "Geese"
)In the above code group = 1:8 can be changed specify any
of the available groups. For example group = 2 would plot
just sampling period 2, or group = c(1,3:7) would plot just
sampling period 1 and 3 to 7.
The command for running the simmr model is identical to
before:
simmr_groups_out <- simmr_mcmc(simmr_groups)or
simmr_groups_out_ffvb <- simmr_ffvb(simmr_groups)simmr will automatically run the model for each group in
turn. This may take slightly longer than a standard single group
run.
The summary command works the same as before. By default
they will produce output for all groups, or you can specify the groups
individually, e.g.:
summary(simmr_groups_out,
type = "quantiles",
group = 1
)
summary(simmr_groups_out,
type = "quantiles",
group = c(1, 3)
)
summary(simmr_groups_out,
type = c("quantiles", "statistics"),
group = c(1, 3)
)For plotting output with multiple groups you can only specify a single group to plot. This is so that you are not overwhelmed with plots:
plot(simmr_groups_out,
type = "boxplot",
group = 2,
title = "simmr output group 2"
)
plot(simmr_groups_out,
type = c("density", "matrix"),
group = 6,
title = "simmr output group 6"
)Whilst you can use the compare_sources function for
multi-group data, there is also an extra function for comparing a single
source between groups via the compare_groups function. This
allows for probabilistic output and plots comparing a single source
across different groups. The simplest use is where you want to compare
just two groups:
compare_groups(simmr_groups_out,
source = "Zostera",
groups = 1:2
)This produces the probability of the group 1 dietary proportion of
Zostera being greater than that of group 2. It also produces a boxplot
of the difference between the dietary proportions and will save this
into a new object if specified. You can turn the plot off by adding the
argument plot = FALSE.
If you specify more than two groups simmr will produce
the most likely probabilistic orderings of the groups as well as the
boxplot as before:
compare_groups(simmr_groups_out,
source = "Zostera",
groups = 1:3
)Combining sources
A common request is that of combining sources. We would recommend
always doing this after running simmr, known as a-posteriori
combining. Suppose for example, you wish to combine the U.lactuca and
Enteromorpha sources which lie in a similar region in the isospace plot
of the Geese data. To proceed, we can create a new simmr
object using the combine_sources function:
simmr_out_combine <- combine_sources(simmr_out,
to_combine = c(
"Grass",
"Enteromorpha"
),
new_source_name = "U.lac+Ent"
)
plot(simmr_out_combine$input)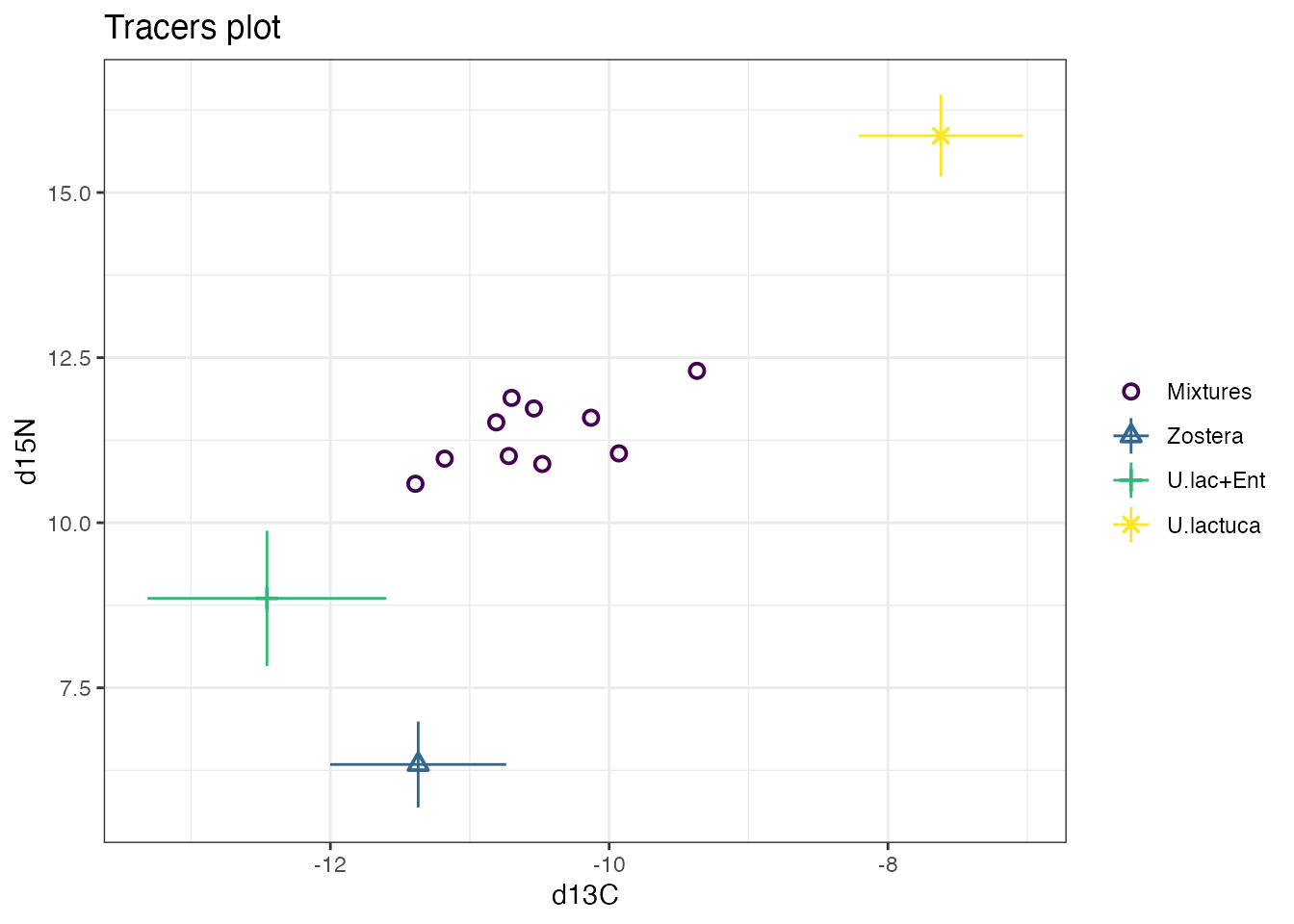
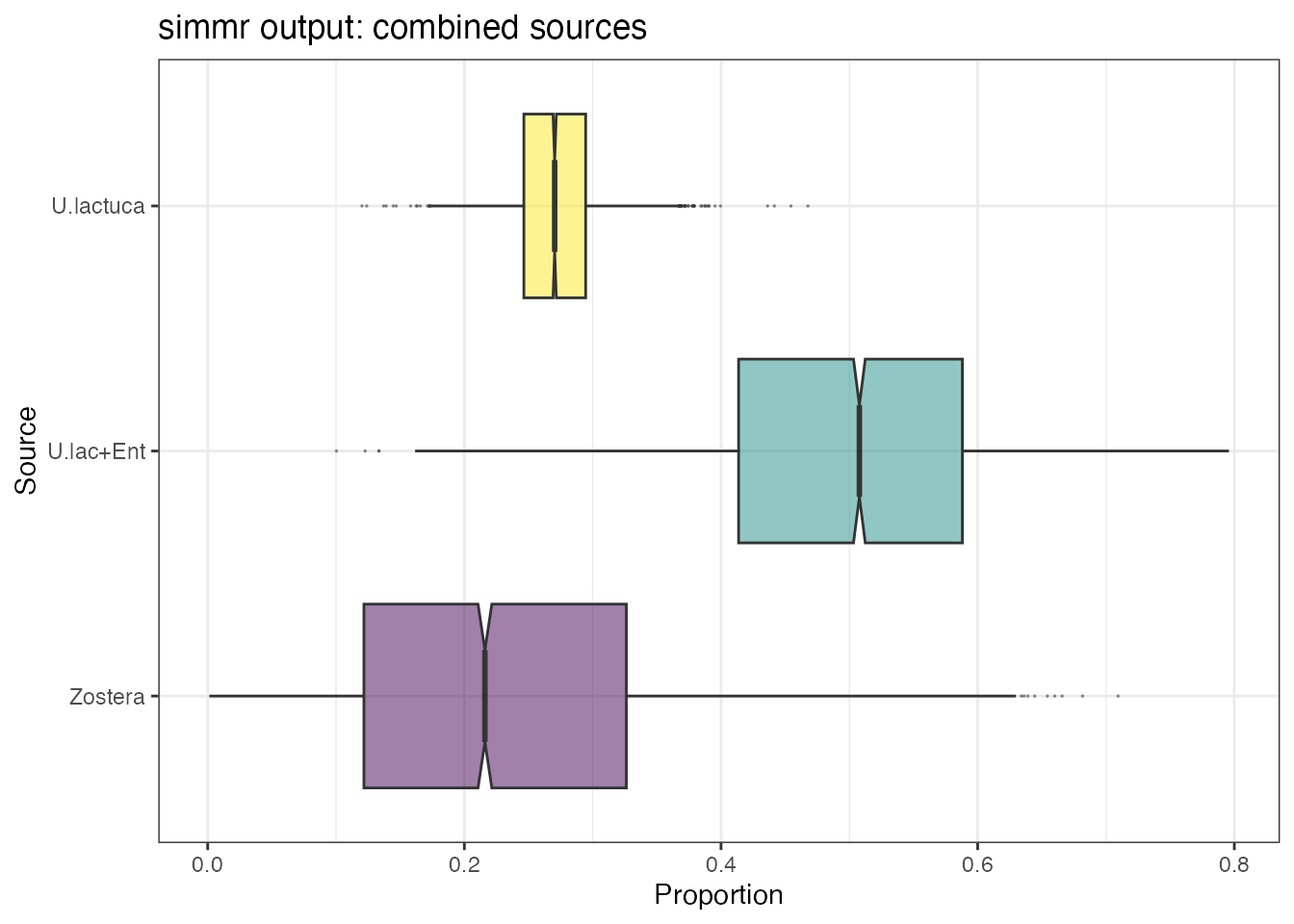
plot(simmr_out_combine,
type = "boxplot",
title = "simmr output: combined sources"
)
This will also work with multiple sources and/or multiple groups:
simmr_groups_out_combine <- combine_sources(simmr_groups_out,
to_combine = c(
"Zostera",
"U.lactuca",
"Enteromorpha"
),
new_source_name = "U.Lac+Ent+Zos"
)
plot(simmr_groups_out_combine$input,
group = 1:8
)
plot(simmr_groups_out_combine,
type = "boxplot",
title = "simmr output: combined sources",
group = 8
)
plot(simmr_groups_out_combine,
type = "matrix",
title = "simmr output: combined sources",
group = 8
)
# And we can now compare sources across groups on this new data set
compare_groups(simmr_groups_out_combine,
source = "U.Lac+Ent+Zos",
group = 1:3
)Running simmr with only one isotope
simmr will run fine with only one tracer, and no changes
should be required to any of the functions. Here is an example with only
one isotope:
mix <- matrix(c(
-10.13, -10.72, -11.39, -11.18, -10.81, -10.7, -10.54,
-10.48, -9.93, -9.37
), ncol = 1, nrow = 10)
colnames(mix) <- c("d13C")
s_names <- c("Zostera", "Grass", "U.lactuca", "Enteromorpha")
s_means <- matrix(c(-14, -15.1, -11.03, -14.44), ncol = 1, nrow = 4)
s_sds <- matrix(c(0.48, 0.38, 0.48, 0.43), ncol = 1, nrow = 4)
c_means <- matrix(c(2.63, 1.59, 3.41, 3.04), ncol = 1, nrow = 4)
c_sds <- matrix(c(0.41, 0.44, 0.34, 0.46), ncol = 1, nrow = 4)
conc <- matrix(c(0.02, 0.1, 0.12, 0.04), ncol = 1, nrow = 4)Now load in with simmr_load:
simmr_in_1D <- simmr_load(
mixtures = mix,
source_names = s_names,
source_means = s_means,
source_sds = s_sds,
correction_means = c_means,
correction_sds = c_sds,
concentration_means = conc
)Create a plot. plot.simmr_input automatically creates a
1D version of these plots:
plot(simmr_in_1D)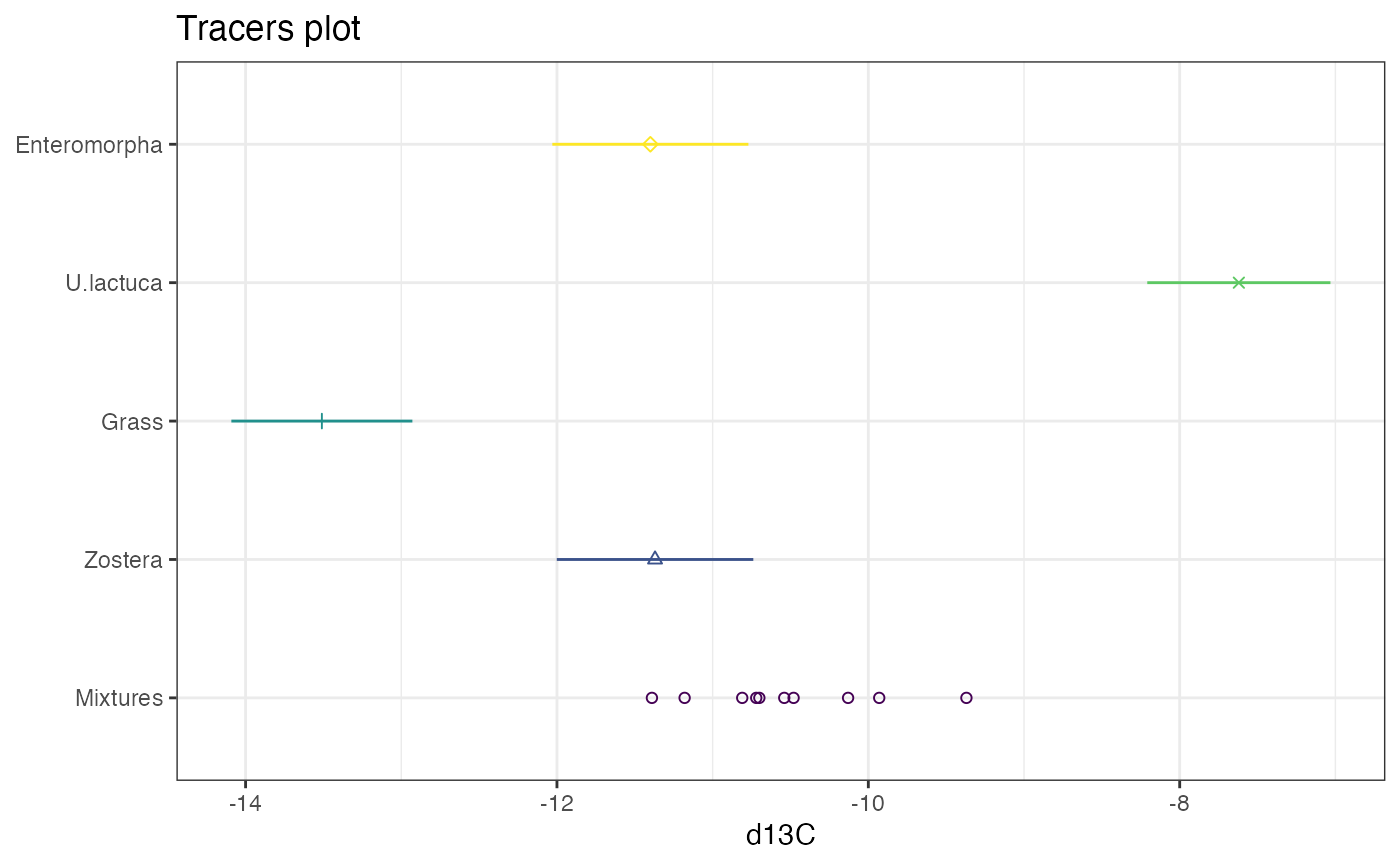
Now run simmr:
simmr_run_1D <- simmr_mcmc(simmr_in_1D)## Compiling model graph
## Resolving undeclared variables
## Allocating nodes
## Graph information:
## Observed stochastic nodes: 10
## Unobserved stochastic nodes: 5
## Total graph size: 83
##
## Initializing modelor
simmr_run_1D_ffvb <- simmr_ffvb(simmr_in_1D)Plot output
plot(simmr_run_1D, type = "boxplot")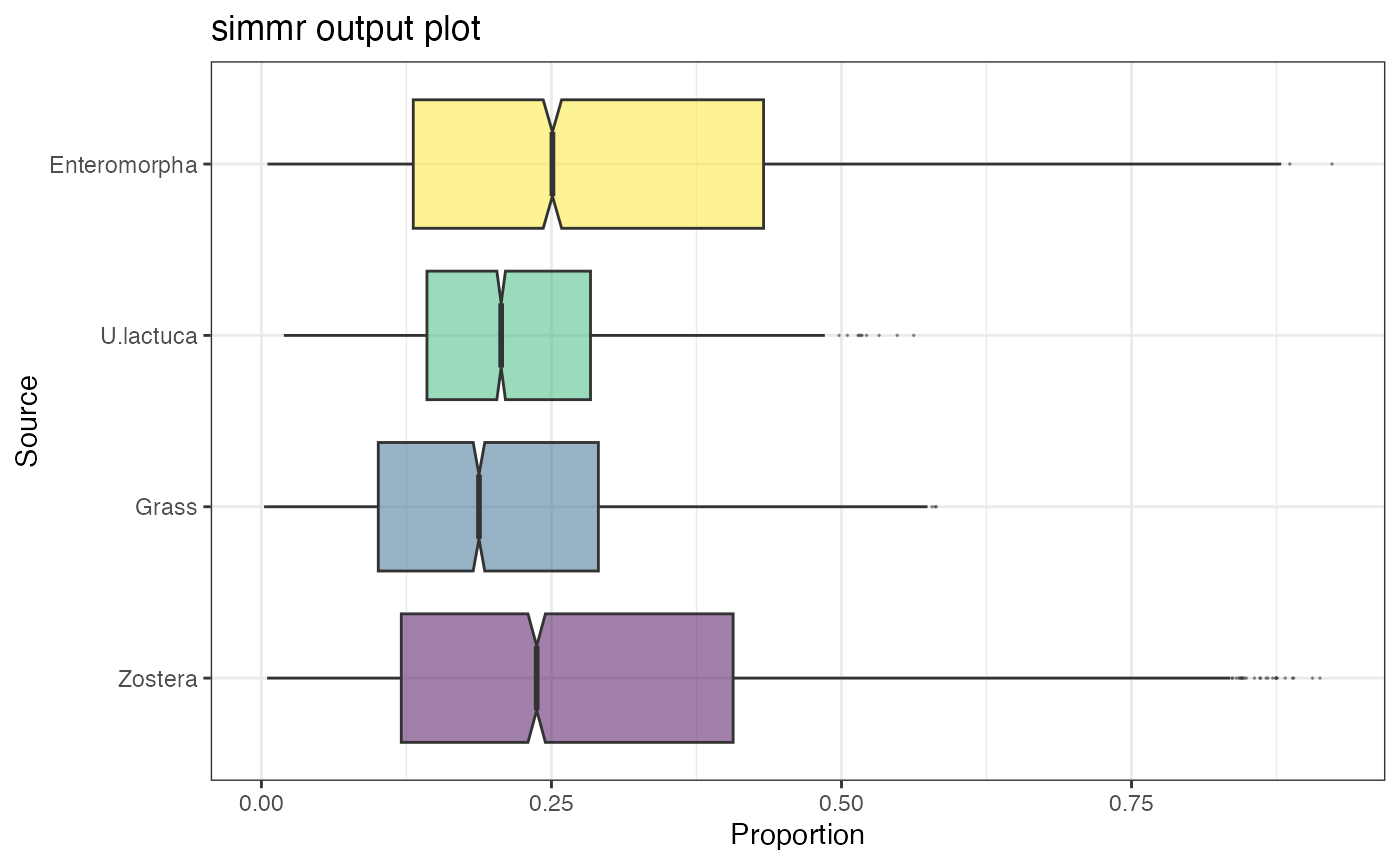
The other summary, compare and
plot functions should all work the same.
Setting up your own prior distributions
Most Bayesian models will work better when you include informative
prior distributions on the dietary proportions. For arguments as to why
you should use prior informative information (and where you could get it
from), see here.
simmr tries to make this easier for practitioners by
including a specific function (`simmr_elicit’) for including prior
information.
We will use the simmr_out object created above. A
reminder about the posterior values
summary(simmr_out, type = "quantiles")All these dietary proportions are very similar. We now suppose we had prior information (e.g. from stomach or fecal contents) that the mean dietary proportions were
proportion_means <- c(0.4, 0.3, 0.2, 0.1)…and proportion standard deviations:
proportion_sds <- c(0.08, 0.02, 0.01, 0.02)We put this into the simmr_elicit function as
follows:
prior <- simmr_elicit(
4, proportion_means,
proportion_sds
)This may take a few moments to run as the code tries to optimise the parameters of a prior distribution which matches these means and standard deviations, which sometimes may not be exactly possible.
When finished, the model can be run using these prior distributions:
simmr_out_informative <- simmr_mcmc(simmr_in,
prior_control =
list(
means = prior$mean,
sd = prior$sd
)
)The new quantiles are:
summary(simmr_out_informative,
type = "quantiles"
)We can plot these priors with their posteriors
prior_viz(simmr_out_informative)Customising plots
Many of the plots in simmr can be customised by adding
on extra options just like a standard ggplot. For
example:
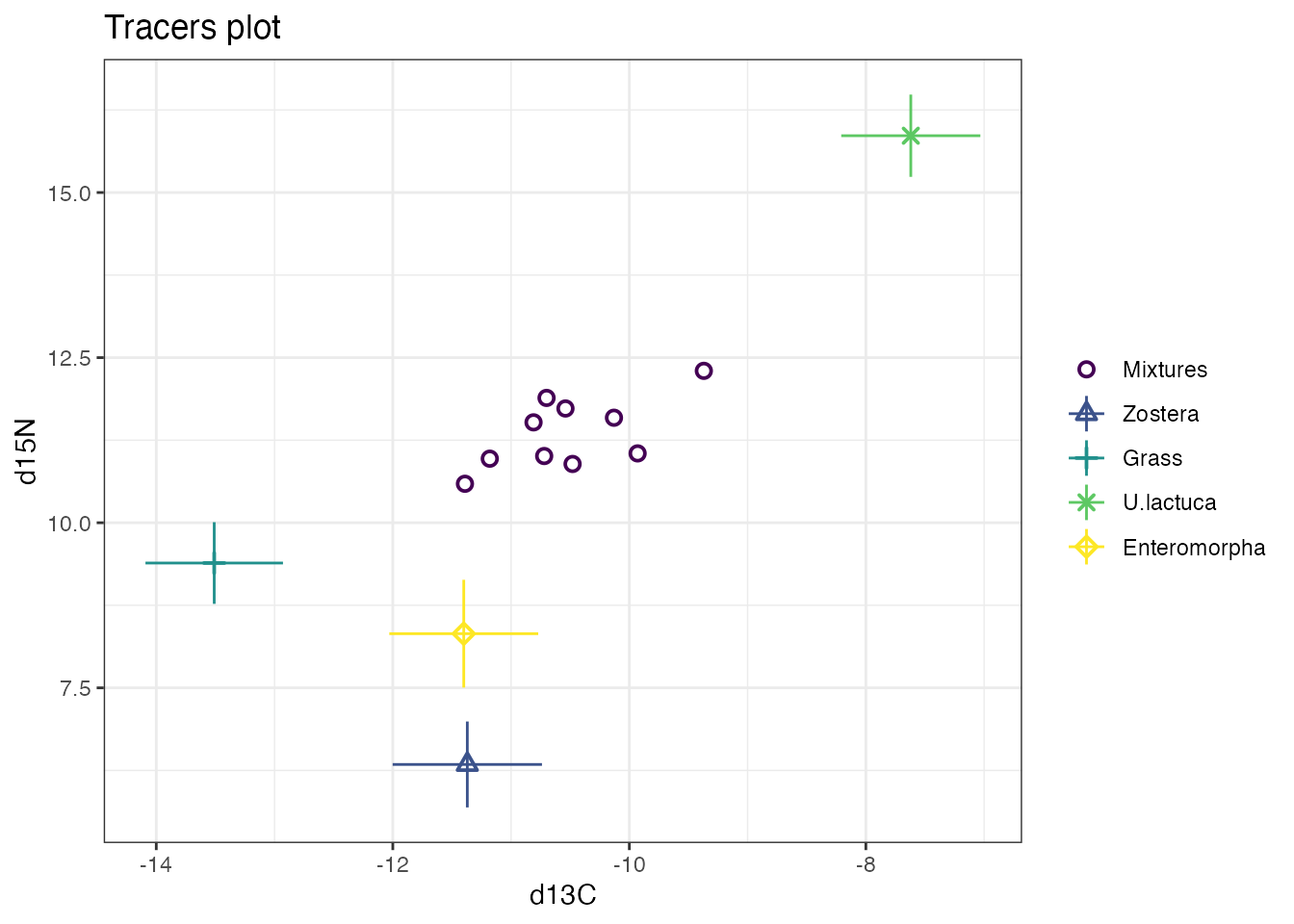
or even on the output:
Note that the above actually changes the x-axis and not the y-axis despite the above command. This is because the coordinates are flipped in the ggplot.
Other options you might like to customise include labels on axes, titles/subtitles, etc. More complicated changes can be made but these involve changing the ggplot commands that simmr uses in the background.
Here is an example where I change the colours of the boxplots:
# First extract the dietary proportions
simmr_out2 <- simmr_out$output[[1]]$BUGSoutput$sims.list$p
colnames(simmr_out2) <- simmr_out$input$source_names
# Now turn into a proper data frame
df <- reshape2::melt(simmr_out2)
colnames(df) <- c("Num", "Source", "Proportion")
# Finally create the new variable that you want to colour by
df$new_colour <- "Type 2"
df$new_colour[df$Source == "Zostera"] <- "Type 1"
# And create the plot
ggplot(df, aes_string(
y = "Proportion", x = "Source",
fill = "new_colour", alpha = 0, 5
)) +
geom_boxplot(notch = TRUE, outlier.size = 0) +
theme_bw() +
ggtitle("simmr output boxplot with changed colours") +
theme(legend.position = "none") +
coord_flip()## Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`.
## ℹ See also `vignette("ggplot2-in-packages")` for more information.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.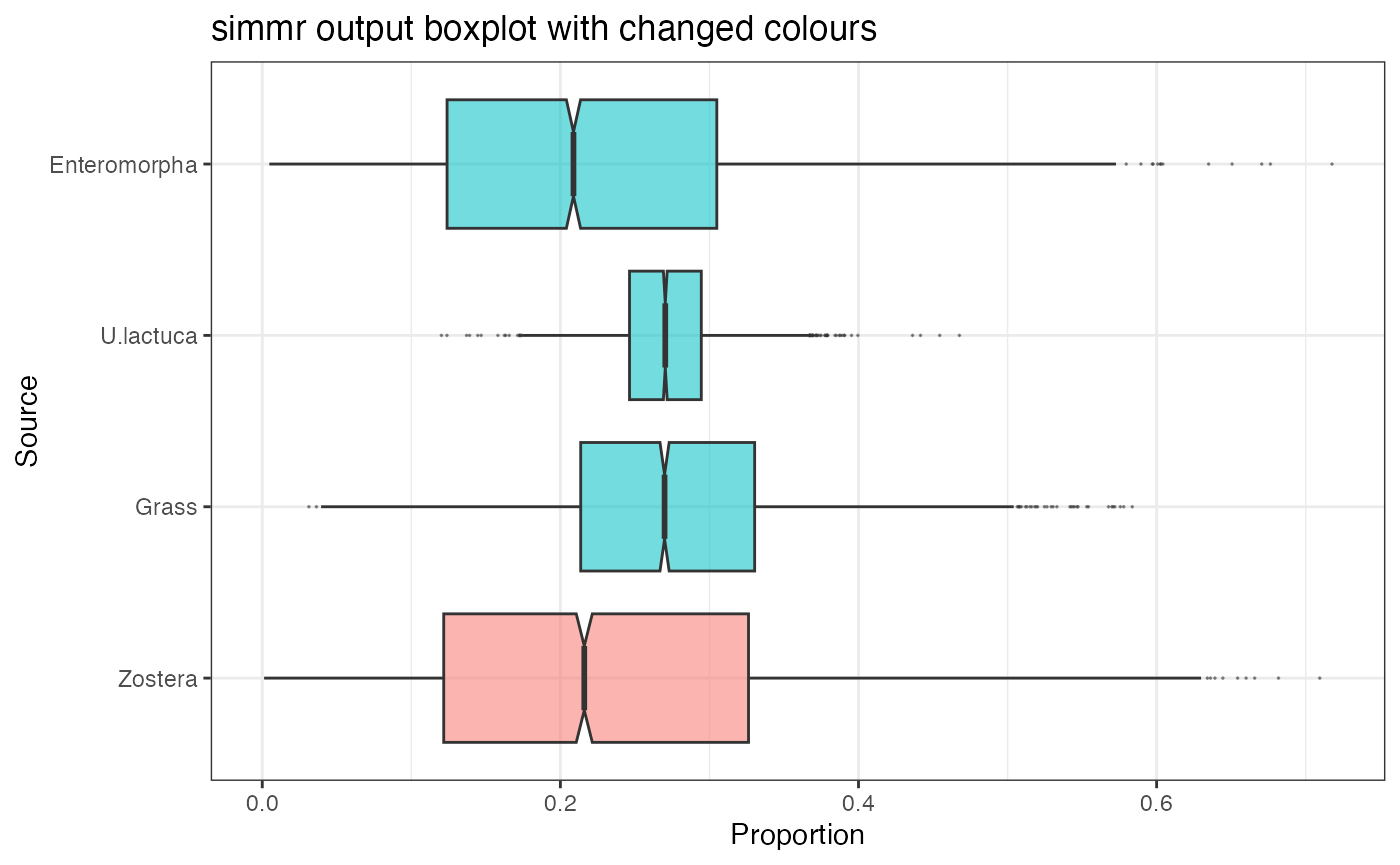
Other advanced use of simmr
Whilst the above gives an introduction to the basic functions of
simmr, the package is open source and all code is open to editing. The
two objects created as part of this vignette simmr_in and
simmr_out are R lists. They can be explored with e.g.
str(simmr_in)which will show their contents. The simmr_out object in
particular allows for full access to all of the posterior dietary
proportion samples. We can calculate for example the mean of the Zostera
dietary proportion on the first (or only) group:
mean(simmr_out$output$`1`$BUGSoutput$sims.list$p[, "Zostera"])## [1] 0.2316519The backquotes around the 1 are required above because they specify which group (the first). We can thus find the probability that the posterior dietary proportion for Zostera is bigger than for Grass:
mean(simmr_out$output$`1`$BUGSoutput$sims.list$p[, "Zostera"]
> simmr_out$output$`1`$BUGSoutput$sims.list$p[, "Grass"])## [1] 0.3919444With more detailed R knowledge, it is possible to create scripts
which run multiple data sets in richer fashions than the default
simmr functions. See the help file
help(simmr_mcmc) for a full list of examples.
Appendix - suggested reading
For the maths on the original SIAR model:
Andrew C Parnell, Richard Inger, Stuart Bearhop, and Andrew L Jackson.
Source partitioning using stable isotopes: coping with too much
variation. PLoS ONE, 5(3):5, 2010.
For the geese data:
Inger, R., Ruxton, G. D., Newton, J., Colhoun, K., Robinson, J. A.,
Jackson, A. L., & Bearhop, S. (2006). Temporal and intrapopulation
variation in prey choice of wintering geese determined by stable isotope
analysis. Journal of Animal Ecology, 75, 1190–1200.
For the maths behind the more advanced JAGS models:
Andrew C. Parnell, Donald L. Phillips, Stuart Bearhop, Brice X. Semmens,
Eric J. Ward, Jonathan W. Moore, Andrew L. Jackson, Jonathan Grey, David
J. Kelly, and Richard Inger. Bayesian stable isotope mixing models.
Environmetrics, 24(6):387–399, 2013.
For some good advice about mixing models:
Donald L Phillips, Richard Inger, Stuart Bearhop, Andrew L Jackson,
Jonathan W Moore, Andrew C Parnell, Brice X Semmens, and Eric J Ward.
Best practices for use of stable isotope mixing models in food-web
studies. Canadian Journal of Zoology, 92(10):823–835, 2014.